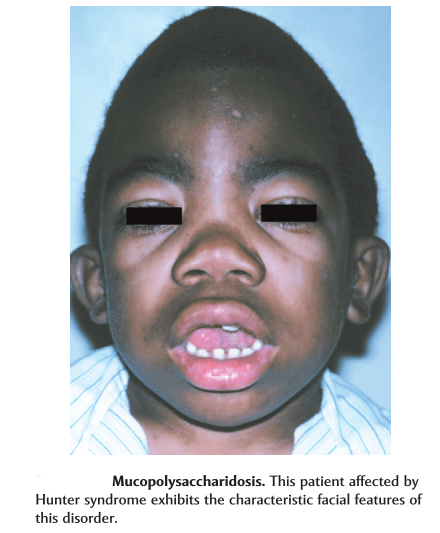
Pigmentation is the process of deposition of pigments in the tissue.Mostly found in the mouth and could be attributed to oral manifestations of systemic diseases or malignancies.This could be due to :
- Increased number of melanocytes
- Augmentation of melanin production
- Deposition of accidentally introduced exogenous materials

The various oral pigmentations can be in the form of:
1)Blue /Purple vascular lesions-
- Hemangioma
- Varix
- Angiosarcoma
- Kaposi’s sarcoma
- Hereditary hemorrhagic telangiectasia
2)Brown melanotic lesions-
- Ephelis & oral melanotic macule
- Nevus
- Malignant melanoma
- Drug induced melanosis
- Physiologic pigmentation
- Cafè au lait pigmentation
- Smoker’s melanosis
- Endocrinopathic pigmentation
- Peutz-jeghers syndrome
- HIV oral melanosis
3)Brown heme associated lesions-
- Ecchymosis
- Petechia
- Hemochromatosis
- Hemorrhagic mucocele
- Thrombosed varix
4)Gray/black pigmentations-
- Silver amalgam tattoo
- Graphite tattoo
- Heavy metal ingestion-lead,mercury,bismuth
- Hairy tongue
SPOTS
Appearance of peculiar spots in specific sites on the body are often diagnostic for major systemic disease or condition or an infection.Some of them are discussed here;
- Koplik spots -measles (rubeola)
- Pink spots on teeth – internal resorption
- Roth spots – subacute endocarditis, typhoid fever
- Bitot’s spots – white plaque on conjunctiva of vitamin A deficient children
- Herald spots -primary lesion seen in pitryiasis rosea
- Sore spots -traumatic ulcers from denture irritation mostly
- Cafè au lait spot-neurofibromatosis, Macune-Albright syndrome,Peutz-jeghers syndrome
Sources: Shafers textbook of oral pathology ,http://www.ncbi.nlm.nih.gov (oral pigmentation : A review),http://www.gibbleguts.com