

Source- textbook of oral pathology Shafers and images from Google
Source- textbook of oral pathology Shafers and images from Google
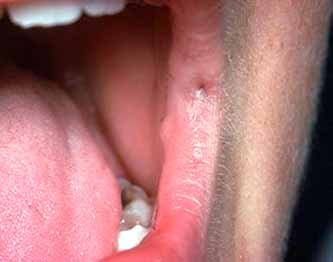
DENTIGEROUS CYST
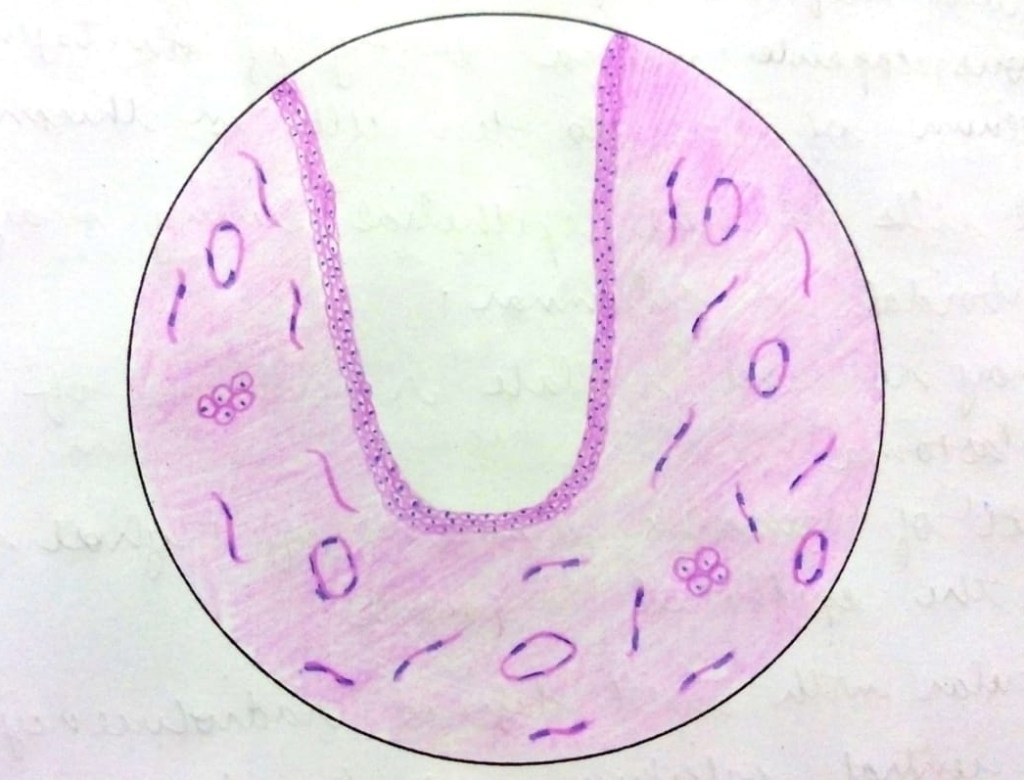
• The lining epithelium is derived from reduced enamel epithelium and hence appears as uniformly thin non-keratinized epithelium.
• The epithelium comprises of 2-3 layers of flattened cells and is characteristically devoid of rete ridges.
Connective tissue capsule is derived from dental follicle, consists of young fibroblasts widely separated by ground substance rich in mucopolysaccharide.
• Connective tissue capsule may show odontogenic epithelial remnants.
• Cystic lumen contains cystic fluid which is thin, watery or may be blood tinged.
ODONTOGENIC KERATOCYST

• Typically shows a thin friable wall which is often difficult to enucleate from the bone in one piece.
• Cystic lumen may contain a clear liquid that is similar to a transidase of serum or it may be filled with a cheesy material, that on microscopic examination consists of keratinaceous debris.
• Microscopically, thin fibrous wall is essentially derated of any inflammatory infiltrate.
• Epithelium may show infoldings into the connective tissue capsule and may be separated from capsule in some areas.
CALCIFYING ODONTOGENIC CYST

• The lining epithelium is of variable thickness commonly around 5-10 layers.
• Spinous cells are loosely arranged.
• Presence of Ghost cells.
• Ghost cells are degenerating cells that appear as void, eosinophilic cells with nucleus showing different stages of degeneration.
• These cells are seen in thickened areas of lining epithelium.
• Connective tissue capsule is often scanty and sometimes contain discrete islands of odontogenic cells.
RADICULAR CYST
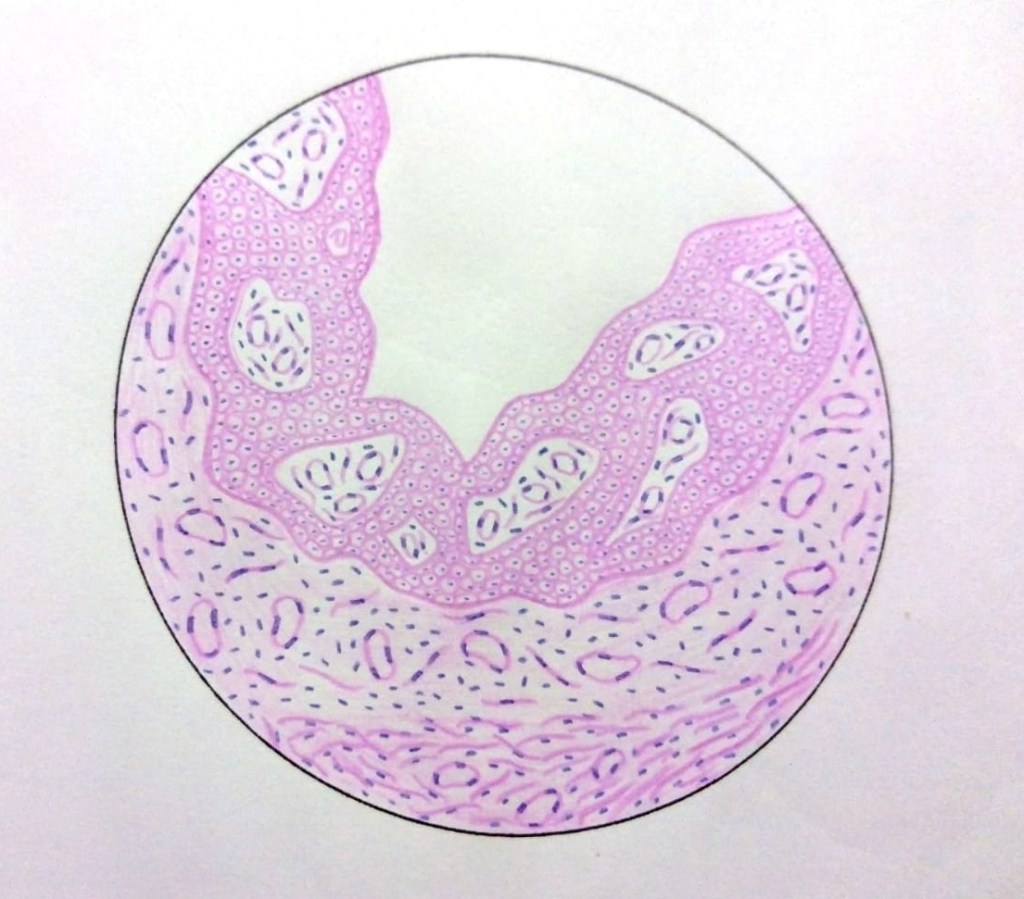
• The epithelial lining of the radicular cyst is stratified squamous in type of variable thickness.
• Epithelium shows spongiogenesis and inflammatory cell infiltration.
• Epithelium may show arc-shaped structures called Rushton bodies.
• The surrounding connective tissue shows granulomatous reaction due to the presence of Cholesterol clefts.
Source: Maji Jose
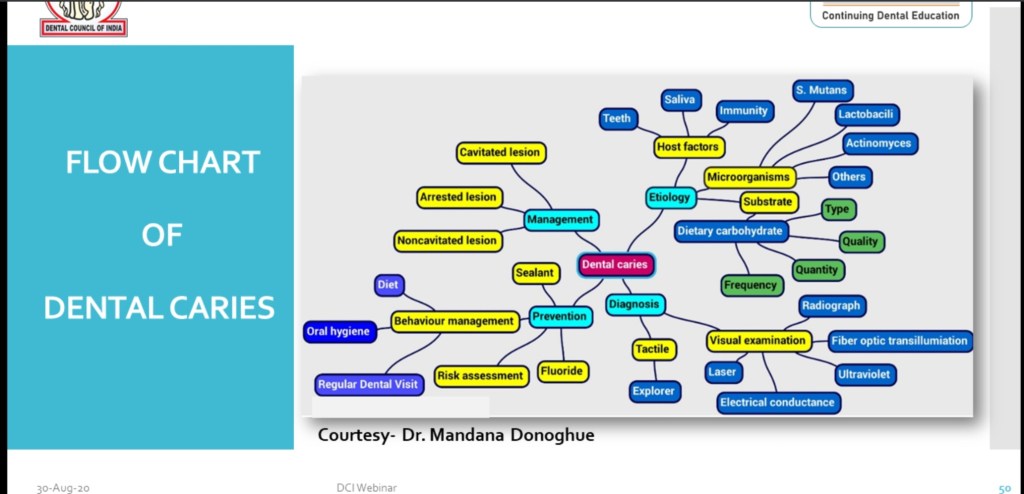
Flow Chart of Dental Caries includes:-
•Etiology
•Diagnosis
•Management
•Prevention
Source:- DCI WEBNEIR on Dental caries-30 Aug 2020.
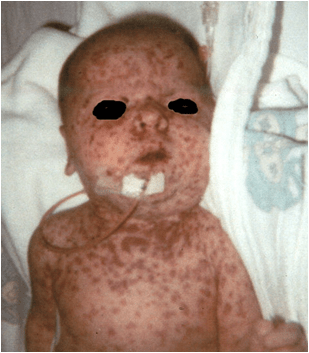
Clinical features:-
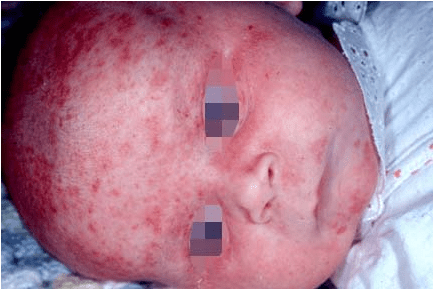
Oral manifestations:-
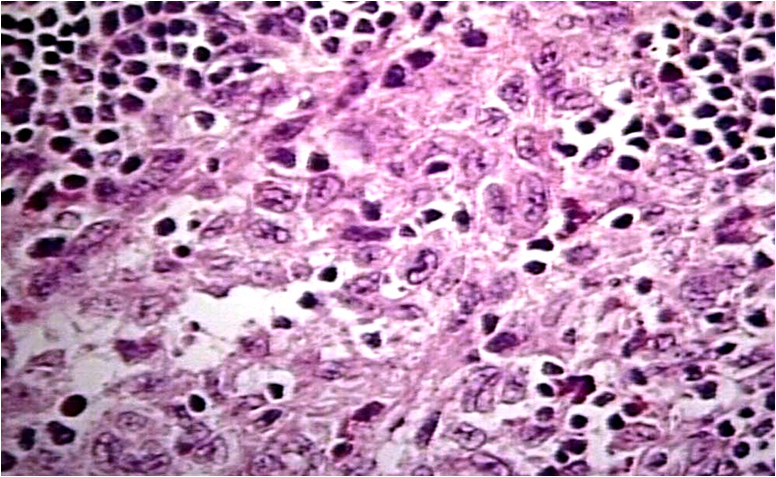
Histologic Features:-
1.Very similar to HSC; histiocytic proliferation with or without eosinophils.
2.These histiocytes do not contain significant amounts of cholesterol.
3.‘Foam cells’ not a feature.
Treatment & Prognosis:-
References:-
Shafer’s 8th edition
PLEOMORPHIC ADENOMA
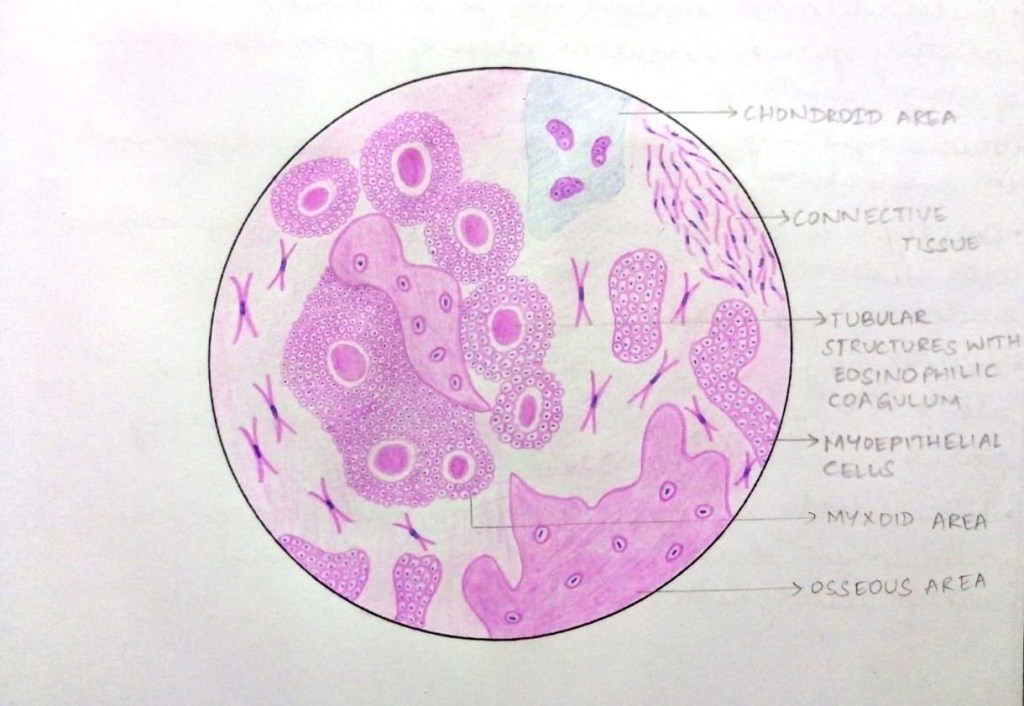
• It is a well circumscribed tumour with complete or partial encapsulation with dense fibrous tissue.
• Epithelial components include proliferating ductal and myoepithelial cells forming ductal structures containing eosinophilic material.
• These cells also may form sheets, strands or islands.
• Ductal cells are cuboidal in shape with scanty cytoplasm.
WARTHIN TUMOUR

• Warthin tumour is made up of epithelial component and lymphoid component.
• Cystic formation, papillary projections are seen, showing germinal centres.
• The cyst are lined by papillary proliferation of bilayered oncocytic epithelium.
• The epithelial cells are abundant, finely granular, eosinophilic and are arranged in two layers.
• The inner luminal layer consists of tall columnar cells with centrally placed, palisaded and slightly hyper-chromatic nuclei.
• The outer luminal layer consists of cuboidal or polygonal cells with more vesicular nuclei.
MUCOEPIDERMOID CARCINOMA

• Microscopically, three types of cells are seen, dispersed in the connective tissue stroma.
• First type is Epidermoid cells which are squamous with distinct intercellular bridges, rarely with evidence of keratin formation.
• Second type is mucous secreting cells which are ovoid, filled with mucin and peripherally placed nucleus (clear cells)
• Intermediate cells are another type of cells, ovoid with darkly staining nucleus and scanty eosinophilic cytoplasm.
ADENOID CYSTIC CARCINOMA

• The tumour is composed of uniform cells resembling basal cells arranged in anastamosing whorls or duct like pattern.
• Some of these duct like areas contain mucoid material.
• This feature gives rise to the characteristic appearance described as “cribriform”, “honeycomb” or “swiss cheese” pattern.
• There may be areas where the cells are tubular or more solid
• The connective tissue components is often hyalinized and surrounding the tumour cells, forming a structural pattern of cylinders (ACC aka cylindroma).
Source: Maji Jose
SQUAMOUS PAPILLOMA

• Papilloma is characterized by finger-like projections lined by hyperplastic stratified squamous keratinized epithelium.
• Each finger like projection has a central thin connective tissue core carrying the blood vessels.
LEUKOPLAKIA

• Histopathologically, leukoplakia is characterized by Hyperkeratosis, Acanthosis and Dysplastic features.
• Dysplastic features include bulbous or drop shaped rete ridges, basal cell hyperplasia, loss of polarity of basal cells, irregular epithelial stratification, cellular pleomorphism, alteration in nuclear cytoplasmic ratio, nuclear hyper chromatism and increased mitosis.
ORAL SUBMUCOUS FIBROSIS

• The epithelium is atrophic with short or flat rete ridges.
• Connective tissue shows juxta epithelial hyalinization and exhibits fibrosis with dense bundles of collagen fibres.
• Focal collections of chronic inflammatory cells are present.
• In severe cases, muscles undergo degenerative changes.
SQUAMOUS CELL CARCINOMA

• The most significant microscopic feature of squamous cell carcinoma is dysplastic epithelial cells invading connective tissue.
• These cells may be arranged in the form of cords, sheets or islands.
• Dysplastic features seen are hyperchromatism of nuclei, alteration of nuclear cytoplasmic ratio, pleomorphism of cells and nuclei, prominent nucleoli, many normal and abnormal mitotic figures, individual cell keratinization and keratin pearl formation.
Source: Maji Jose
Clinical features:-
Oral manifestations:-

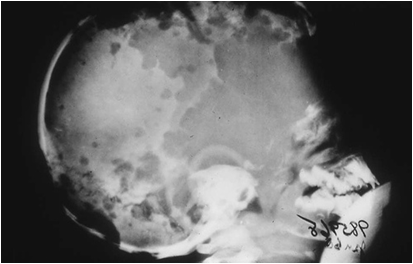
Radiological features:-


Histological features:-

Treatment & prognosis:-
Reference:
1.Faculty notes
2.Google

Clinical Features:-
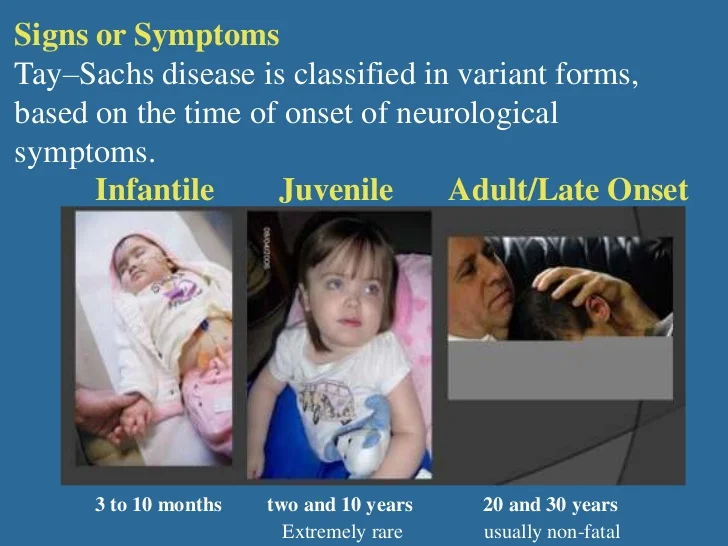
References:-
1.Shafers’ 8th edition
2.Google images
3. For better understanding: https://youtu.be/2z3nSnBe8Vg
Clinical Features:-
Niemann-Pick disease type A begins in the individual’s first few months of life. Symptoms include the following:
◦Feeding difficulties
◦Abdominal enlargement within 3-6 months
◦Progressive loss of early motor skills
◦Rapid decline leading to death by the time the patient is aged 2-3 years
◦Abdominal enlargement may be detected in early childhood.
◦Respiratory infections recur.
◦No neurologic involvement is present.
Symptoms may include the following:
◦Unsteadiness of gait, clumsiness, problems in walking
◦Difficulty in posturing of the limbs
◦Slurred, irregular speech
◦Learning difficulties and progressive intellectual decline
◦Sudden loss of muscle tone, which may lead to falls
◦Seizures
Tremors accompanying movement.
Histologic features:-
Niemann – Pick cells are foamy, lipid – laden cells distributed throughout RES.
Positive for cholesterol & only weakly positive for ALP.
Affected cell becomes extremely large, enlarged secondary to distention of lysosomes.
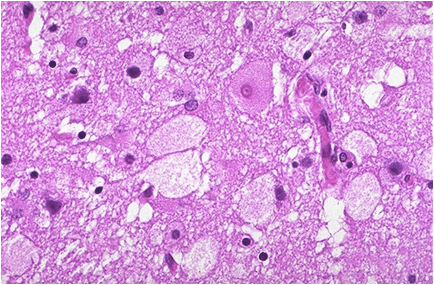
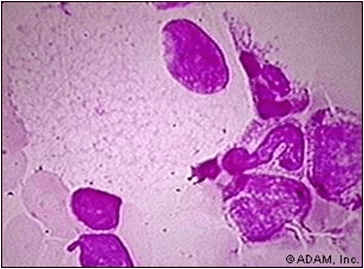
Treatment:-
Enzyme replacement therapy currently being explored.
Current treatment symptomatic; consists mainly of antibiotic therapy for infections of pulmonary involvement.
Organ transplant (liver) also proposed
Overall prognosis poor.
References:-
Shafers’ 8th edition
Common LSD, characterized by deposition of gluco – cerebroside in cells of macrophage – monocyte system.
Results from mutation in gene or deficiency of enzyme that codes for glucosylceramidase.
Leads to accumulation of glucosylceramide in mononuclear phagocytic cells; transformed into “Gaucher cells“.
Five autosomal recessive variants exist resulting from distinct allelic mutations.
Three have been described in the literature.
Pathogenesis:-
These transit through blood as macromolecules; engulfed by phagocytic cells of the body.
Type I – Chronic nonneuronopathic form:-
Characterized by clinical or radiologic bone involvement.
Spleen enlarges massively filling the entire abdomen.
Type II – Infantile/acute neuronopathic form:-
Rapidly progressive neurovisceral involvement.
Symptoms start before 2 years of age, very severe.
Results in death in infancy.
Type III – Juvenile/ Norrbotnian form:-
Patients are juvenile presenting with systemic involvement.
(intermediate between type I and type II).
Progressive CNS involvement usually begins in teens or early twenties.
Histologic Features:-
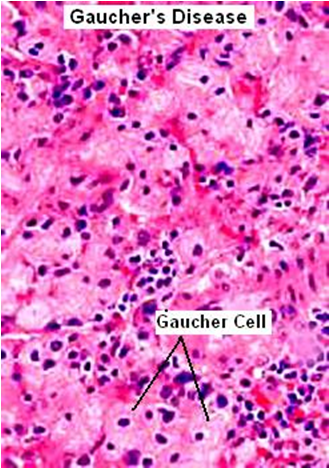
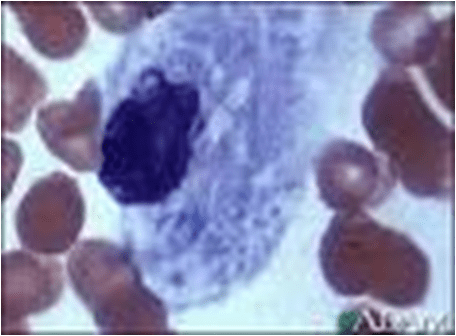
Treatment & Prognosis:-
Prognosis of Type II is very poor; death within first year.
Less virulent form may persist till 6th decade.
Administration of purified glucocerebrosidase results in dramatic decrease in hepatic accumulations of glucocerebroside.
Enzyme replacement therapy available; effective but extremely expensive.
REFERENCES:-
Shafer’s 8th edition
