- General formula of carbohydrate = (CH20)n
- Following are monosaccharides
- Glucose
- Fructose
- Galactose
- NON REDUCING SUGARS = trehalose
- REDUCING SUGAR = Maltose, lactose and cellobiose
- Pentose sugar present in heart cells = lyxose
- Monoscaarchirde glucose forms a part of the disaccharide sucrose
- GALACTOSE
- not an oligosaccharide
- Monosaccharide with a maximum rate of absorption in intestine
- SUCROSE
- sugar characterised by its non-reducing property. It is also called cane sugar and table sugar
- It cannot be metabolised
- Invertase catalyzes the hydrolysis of = Sucrose into fructose and glucose**
- FRUCTOSE
- In hereditary fructose intolerance, one can see intolerance to = Fructose and Sucrose
- A patient with hereditary fructose intolerance is deficient in = Aldolase enzyme
- Highest concentrations of fructose are found in = Seminal fluid
- LACTOSE
- Milk sugar
- Hydrolysis of lactose yields glucose and galactose
- Beta 1.4 glycosidic bond is present in = Lactose
- GLUCOSE
- Glucose, Maltose and Lactose = Exhibit inversion
- Malatose
- is dissacride of = Glucose and glucose
- Glycosidic linkage = alpha 1 – 4
- Epimeric Pair = D Glucose and D Galactose
- Glucose monomers in glycogen are held by = Alpha 1-4 bonds, alpha 1-6 bonds
- Only sugar absorbed against concentration gradient = Glucose
- True blood sugar level measures the levels of = Glucose + Fructose
- Abnormal constituents of urine = Ketone + Glucose
- Renal threshold of glucose = 180 mg/dl
- In prolonged starvation, the main energy source of brain = Ketone bodies
- GLUCOSE can be synthesised from = Glutamate, Asparate and Alanine
- The uptake of glucose by the liver increases followed by carbohydrate meal because = increase in phosphorylation of glucose by glucokinase
- Apart from liver, glucokinase is present in = Pancreatic islet cells
- Glucokinase = It is a Inducible enzyme
- In glucose solution at equilibrium, mutarotation results in a fixed optical rotation of = 52.5*
- Form of glucose predominatly seen is as = Beta D Glucopyranose
- Glycemic index is highest for = Glucose
- Amount of asymmetric carbon atoms in glucose = FOUR
- Number of stereoisomers of glucose is = 16
- Substrate used by RBC in fasting state = Glucose
- Important precursors of glucose in animals
- Pyruvate
- Lactate
- Glycerol
- Major factor that determine weather glucose is oxidized by aerobic or anaerobic glycolysis = NADH and ATP/ADP Ratio
- Potassium Oxalate and Sodium Fluoride = added to the blood sample to estimate glucose
- Muscle cannot release glucose from glycogen because of the deficiency of = Glucose 6 phosophatase
- GLYCOGEN
- Starch and Glycogen are polymers of = Alpha Glucose
- Amylose and amylopectin are constitutes of = Starch
- CARBOHYDRATE RESERVE OF THE BODY = glycogen
- Glycogen breakdown = Glucose and Lactic acid
- Tissue with highest glycogen content = Liver
- An essential for the conversion of glucose to glycogen in the liver = UTP
- Muscle glycogen is mainly utilized for supplying energy to = Liver
- Ditery fiber is rich in = Cellulose
- The rate of absorption of sugars is highest for = Hexoses
- Proteins and carbohydrates of glycoproteins are held by = Glycosidic bonds
- Sialic acids are acetylated derivatives of = Neuraminic acid
- GLYCOLYSIS
- The oxidation of glucose or glycogen to pyruvate and lactate by the EMF pathway is called = Glycolysis
- EMF pathway reaction takes place outside the mitochondria
- Phosphofructokinase is the key rate-limiting enzyme of = Glycolysis
- During the conversation of glycerol to pyruvic acid, the first glycolytic intermediate to form = Dihydroxy acetone phosphate
- Allosteric inhibition with ATP effects and rate-limiting enzyme is = Phosphofructokinase**
- The enzyme involved in the first committed step of glycolysis = Hexokinase
- Enzyme phosphofructokinase 1 is strongly activated by = Fructose 2,6 bisphosphate
- Enzyme Phosphofructokinase is allosterically inhibited by = Citrate
- Whenever the cells ATP supply is depleted = Phosphofructokinase enzyme activity is increased
- ATP is produced by the following enzyme = Pyruvate Kinase
- Glycolysis enzyme inhibited by fluoride is = Enolase
- ENOLASE = Catalyzes the reversible degradation of 2 – phosphoglycerate to phosphoenolpyruvate
- For glucose estimation in blood, the mode of transportation from primary health care to the laboratory is done by the addition of = Sodium Fluoride
- Insulin acts on which enzyme = Glucokinase
- Inhibition of glycolysis in the presence of oxygen is called as = Pasteur Effect
- The reverse of Pasteur effect = Crabtree effect
- The main pathways of metabolism in the brain are = Glycolysis and Citric Acid Cycle
- The end product of glycolysis under anaerobic conditions is = Lactic acid
- Number of ATP molecules generated in the conversion of glycogen to lactate = 2
- Following exercise, the level of lactic acid in blood during ventilation = Decreases
- Ion which is important in the glycolysis = Magnesium
- Pyruvate dehydrogenase and Alpha-ketoglutarate dehydrogenase = Multienzymee complex
- Pyruvate Dehydrogenase complex contains = NAD, FAD, Co-A
- The enzyme which provides a link between glycolysis and citric acid cycle = Pyruvate Dehydrogenase
- Increase in pyruvate and lactate is seen in = Thiamine deficiency
- Unique by product of lycolysis in RBC = 2,3 biphosphoglycerate
- Glycerol enters glycolysis via = Dihydroxyacetone phosphate
- Glycolytic pathway is located in = CYTOSOL
- Glycolysis is always anaerobic in = Erthrocytes
- Enzyme responsible for the conversion of glucose 1 phosphate to glucose 6 phosphate = Phosphoglucomutase
- Irreversible step of glycolysis involves
- Phosphofructokinase
- Pyruvate kinase
- Hexokinase
- True statements regarding anaerobic glycolysis
- End product is lactic acid
- Net production of ATP is 2
- Occurs in places like eyes, lens and RBC’s
- In the glycolysis cycle, substrate-level phosphorylation takes place in = pyruvate kinase
- Generation of ATP upt pyruvate in aerobic glycolysis = 7
- Cancer cells derive energy mainly from = Glycolysis
- CITRIC ACID CYCLE
- Enzymes concerned with the citric acid cycle are found in = Mitochondria
- Kreb cycle occurs in = Aerobic conditions
- Kreb cycle doesn’t occur in = RBC due to absence of mitochondria
- In the TCA cycle, what is formed first = Citrate
- In the TCA cycle, citrate is immediately converted into = Cisaconitate, after losing molecule of H20
- Acid formed in = Oxaloacetic acid
- The correct sequential order of enzymatic reaction of kreb cycle when molecule acetyl-CoA enters the cycle = Citrate, ketoglutarate and oxaloacetate
- In the TCA cycle, substrate-level phosphorylation takes place in = Succinyl CoA to succinate
- The enzyme involved in substrate-level phosphorylation = Succinyl CoA Synthetase
- In the TCA cycle, substrate-level phosphorylation occurs at = Thiokinase
- High energy phosphate compound that is formed via substrate-level phosphorylation = GTP
- Succinyl CoA to Succinate = 1 ATP
- 1 molecule of glucose forms = 2 molecules of pyruvate
- 1 molecule of acetyl CoA enzyme gives rise to = 12 ATP
- Alpha Ketoglutarate = Metabolite which is used in the detoxification of ammonia in the brain
- Acetly CoA can be converted into
- Fatty acids
- Cholesterol
- Ketone bodies
- Total Number of dehydrogenase = 4
- Final common pathway for the oxidation of carbohydrate, lipids and protein in human body is = TCA cycle
- INSULIN
- Glycogen synthesis is increased by = Insulin
- Insulin increases the following pathway
- Glycogen synthesis
- Fatty acid synthesis
- Protein synthesis
- Insulin causes lipogenesis by
- Increasing acetyl CoA carboxylase activity
- Increases the transport of glucose into the cells
- Decreases the intracellular cAMP level
- Glucose transporter which is stimulated by insulin is located in = Skeletal muscle and adipose tissue
- Activity of which of the following enzyme is not affected by insulin = Hexokinase
- Insulin = not polymer of glucose
- Homopolysaccharide made up of fructose is = Insulin
- GLYCOGENEIS
- It requires
- Uridine diphosphate
- Glycogen synthetase
- Branching enzyme
- It requires
- GLYCOGENOLYISIS
- Hypoglycaemia is corrected by increasing the rate of live glycogenolysis = GLUCAGON
- The first product of glycogenolysis = Glucose 1 phosphate
- Rate limiting step of glycogenolysis = Glycogen Phosphorylase
- A deficiency of Phosphorylase = would impair the body’s ability to maintain blood glucose concentration during the first 24 hours
- The conversation of glucose 6 P to G 1 P = example of isomerization
- Adrenaline acts on the enzyme = phosphorylase
- Glycogenolysis in muscle does not raise blood sugar due to lack of = H-6-phosphatase
- Glucose can be synthesized from
- Tryptophan and Phenylalanine
- Glycerol
- Lactic acid and propionic acid
- GLUCONEOGENSIS
- Mainly occurs in = Mainly in Liver and partly in Kidney, not in muscles**
- Liver and Muscle = involved in Cahill Cycle
- Substance for gluconeogenesis = Glycerol
- The key enzyme of gluconeogenesis = Pyruvate carboxylase
- The compound that can give rise to glucose by gluconeogenesis = Lactate
- Amino acids that enter TCA cycle for gluconeogenesis = Phenylalanine, Tyrosine and Tryptophan
- Glycerol is converted to glucose in = Liver
- Major contribution towards hepatic gluconeogenesis = Lactate
- Hepatic gluconeogenesis is stimulated by = Glucagon and Epinephrine
- X = Aspartate
- Y = Oxaloacetate
- Major contribution towards gluconeogenesis is by = Alanine and Glutamine
- Malate shuttle – is important in = Gluconeogensis and Glycolysis
- Substrates of gluconeogensis =
- Glucogenic amino acids
- Lactate
- Glycerol
- HMP Pathway
- TRUE STATEMENTS
- HMP shunt is an alternative pathway for oxidation of glucose that occurs in the cytosol
- It is characterized by the absence of production of ATP
- It is active in the adipose tissue, liver and gonads
- Oxidative phase generate NADPH
- Non oxidative phase generates ribose precursors
- Sites where HMP shunts can occur include
- Liver
- WBC
- Lactating mammary gland
- Testes
- Step in HMP pathway requiring TPP = Transketolase
- HMP shunt is of great importance in cellular metabolism because it produces = NADPH
- Dehydrogenases of HMP Shunt are specific for = NADP
- NADPH is the product of = HMP pathway
- Enzymes which used NADP as coenzyme = Glucose 6 phosphate dehydrogenase = regulartory enzyme in HMP shunt
- First pentose formed in HMP shunt = Ribulose 5 phosphate
- Metabolites in HMP shunt are
- Sedoheptulose 7 phosphate
- Glyceraldehyde 3 phosphate
- Xylulose 5 phosphate
- TRUE STATEMENTS
- GLYCOGEN STORAGE DISORDERS
- Glucose 6 phosphate deficiency
- is seen in = Von Gierke’s disease = autosomal recessive
- Defective cori cycle and increased mobilization of glycogen from liver is seen
- Hyper-ureicemia is feature of = type 1 glycogen disorder (Von Gierke’s disease)
- Type 2 glycogen disorder is due to deficiency of =
- 4 and 1,6 – glucosidase
- lysosomal alpha 1
- McArdles disease is due to deficiency of = Myophosphorylase (Type 5)
- Beta Galactosidase is deficient in = Krabbe’s disease
- Limit detrin accumulate in cytosol = Type 3 – Cori’s disease**
- Pompe’s disease is due to defieceny = Acid Malatase
- Glucose 6 phosphate deficiency
- Galactosaemia commonly is due to deficiency of = galactose 1 phosphate uridyl transferase
- Cytochromes are = iron containing porphyrins
- Main enzyme responsible for the activation of xenobiotics = Cytochrome P-450
- Most lipogenic = fructose
- Xylitol is = Natural five carbon sugar
- Glucose transporters present in the Beta cells of the islets of langerhans is = GLUT 2
- GLUT 4
- Is present in adipose tissue
- Facilitates diffusion of glucose
- Transferred from cytosol to the cell membrane by insulin
- Glucose transporter in myocyte stimulated by insulin is = GLUT4
- Prolonged carbohydrate deficiency leads to = Ketoacidosis
- Glutathione
- Tripeptide
- Conjugates xenobiotics
- Co factor of various enzymes
- Sphingosine is present in = Ceramide
- Haworth structures refer to = Pyran and Furan forms of sugars
- Glycosaminoglycan present in cornea as = Keratan Sulphate and Dermatan Sulphate
- Enantiomers – non superimposbale images of one another
- D mannose and L mannose
- D Glucose and L glucose
- Specific rotation of beta D glucopyranose = +19*
- Hetropolysccaride among the following = Heparin
- Lactate formed in muscles can be utilized through = Cori cycle
- Alpha Lactone is = Vitamin C
- Most prominent carbohydrate component of hemicelllulose = Arabinoxylan
- Rothera’s test is for = Ketones
- Bacterial Glutamine synthethase = Enzyme catalyzes a reaction in which reduced nitrogen is introduced into cellular metabolism
- When velocity of enzyme activity is plotted against substrate concentration = HYPERBOLIC CURVE
- Glycoproteins = are important for white blood cell recognition
- Cytochrome C = recieves flavoproteins
- Anabolism and Catbolism are chemically linked in the form of = ATP
- Factors determining the activity of an enzyme
- Association with regulatory protein
- Sequestration
- Allosteric regulation
- 6 – phosphogluconate dehydrogenase = catalyses the first step in the pentose phosphate pathway
- Flux control coefficient = measure of the effect of an enzyme’s concentration on flux through a multi enzyme pathway
- Frutose 2,6 bisphosphate = compounds are responsible for the coordinated regulation of glucose and glycogen metabolism
- Elasticity Coefficent = measure of how responsive the enzyme is to changes in the concentration of metabolite
- After Overnight fasting, levels of glucose transporters are reduced in = Adipocytes
- Isomerase = enzyme that catalyzes the conversion of an aldose sugar to a ketose sugar
- Insulin stimulated glucose uptake takes place in via GLUT 4 which is insulin dependent
- Heart
- Skelteal Muscle
- Adipose tissue
- Transport of glucose in Liver = GLUT 2 = Insulin independent
- Decrease BMR is seen in = Starvation
- Caloric Value of Alcohol = 7Kcal/g
Category: Biochemistry


Sign and symptoms of acromegaly [ mnemonics]

antihypertensive drugs


Common Tests/Investigations
Here’s a list of the few laboratory tests and investigations which are helpful & specific for diagnosis of certain diseases and conditions.


Sources : Burkets oral medicine 11th edition,Shafer’s textbook of oral pathology 7th edition,www.glasbergen.com
Enzymes Overview
ENZYME CHARACTERISTICS
- Increase rate of reaction by lowering activation energy
- Most are proteins
- Specific – conversion of one specific substance to one product
- May require cofactors or coenzymes
- Carefully regulated
ΔG = free energy
- Free energy of the product minus the free energy of the reactants
- ΔG is negative for enzymatic reaction because energy is released (exergonic reaction)
ΔEa = activation energy
- Energy barrier that must be overcome for a reaction to proceed
- Enzymes lower activation energy of a reaction by stabilizing transition state
- Energy required to get to equilibrium (rate of forward and reverse reactions are the same) correlates with ΔG and is unchanged in the presence or absence of enzyme
- Enzyme doesn’t dictate whether reaction will proceed but determines speed of reaction
ENZYME ACTIVE SITE
- 3D structure produces active site
- Shaped so that substrate fits in
- Product of an enzymatic reaction has lower affinity for binding site: exits binding site and is released
COFACTORS AND COENZYMES
- Bind cofactor binding site (distinct from active site)
- Some enzymes inactive without cofactor or coenzyme
- Many are vitamin-derived, metal ions, or other smaller organic molecules
Urea Cycle
By Dr Musaddika Shaikh Dentowesome @drmusaddikashaikh
Urea cycle :-
- It is end product of protein metabolism.
- It is also known as urea Cycle or ornithine cycle or Krebs hensleit cycle.
- Nitrogen of amino acids get converted to ammonia which is toxic to body.
- Ammonia gets converted into urea and detoxified.
- Urea is mainly synthesized in liver and transported to kidney for excreation.
- Urea synthesis in 5steps cyclic process with 5 distinct enzymes.
- First two enzymes – mitochondria and Rest – cytosol
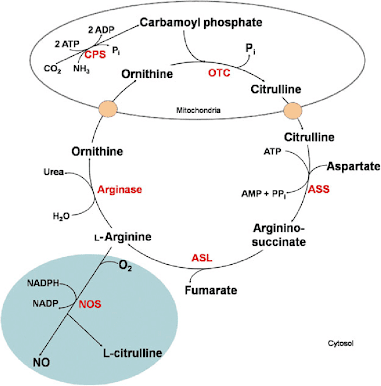
Formation of Carbamoyl Phosphate :- Carbamoyl Phosphate synthase 1st present in mitochondria catalyses condensation of NH ions with CO2 to form carbamoyl phosphate. This step consumes 2ATP and is irreversible.
Formation of citrulline :- It is synthesized from carbamoyl phosphate synthetase 1st and Ornithine by transcarbomyle. Ornithine is regenerated and used in urea cycle.
Formation of Arginosuccinate :- It condenses citrulline with aspartate to produce Arginosuccinate. 2amino group of urea is involved in reaction. This step requires ATP which is cleaved to AMP.
Formation of Argentine :- Arginosuccinate breakes to form argenine and fumarate.
Formation of Urea :- Arginase is final enzyme which cleaves argenine to yeild urea and orthinine. Orthinine enters mitochondria for its reuse in urea cycle.
Reaction :-NH4+CO2+ASPARTATE+3ATP = UREA+FUMARATE+2ADP+2PIi+AMP+Ppi
Reference :- Biochemistry book U. Satyanarayan, U Chakrapani. Google website
Glycolysis
By Dr Musaddika Shaikh Dentowesome @drmusaddikashaikh
Glycolysis :- It is sequence of reaction converting glucose into lactose and pyruvate with the production of ATP
- It is also know as EMBDEN MEYENHOFF Pathway.
- It takes place in cell of body.
- The enzyme are present in cytosomal fraction.
- It occurs in presence of oxygen (aerobic) and absence of oxygen (anaerobic).
- Lactate is end product of anaerobic pathway.
- Pyruvate is end product of aerobic pathway forming CO2 and H2O.
- It is major pathway for synthesis of ATP.
- It is essential for brain.
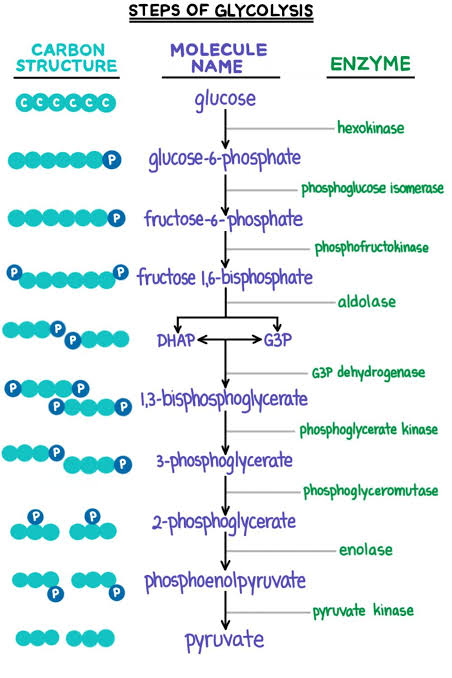
Reaction :- Glucose+2ADP+2Pi = 2Lactase+2APP
Significance :-
- It is formed by active skeletal muscle major precursor for gluconeogenesis
- Lactate is dead end in glycolysis
- It is carried from skeletal muscle through blood and handed over to liver , where it is oxidized to Pyruvate
Reference :- Books Human Physiology for bds A.K Jain Google website expii
MOA OF LOCAL ANAESTHESIA

source – don’t remember, had written it a lot time ago
Phenylalanine & Tyrosine Metabolic Disorders
PHENYLKETONURIA
Pathophysiology: Toxic Metabolites of Phenylalanine
To understand the pathophysiology of phenylketonuria, show that when phenylalanine accumulates at toxic levels, it transaminates into:
- Phenylpyruvate (aka phenyl ketone); hence, “phenylketonuria” describes the presence of phenylpyruvate, phenylalanine, and two key other derivatives in the urine and blood:
- Phenylacetate which has a distinct “must/mousy odor”.
- Phenyllactate.
Phenylalanine Excess / Tyrosine Deficiency
- Thus, overall indicate that in phenylketonuria, there is an:
- Excess of phenylalanine
- Deficiency of tyrosine
So the goal of therapy is to reduce phenylalanine intake and to supplement tyrosine deficiency via the diet. Remember the sparing action of tyrosine on the requirements of phenylalanine
Clinical Presentation of PKU
- Hypopigmentation
- Indicate that hypopigmentation (of the skin and iris) is a finding in this disorder (remember: melanin is a derivative of tyrosine and tyrosine is deficient in PKU).
- Neuropsychiatric disorder
- And because the toxic levels of phenylalanine and its derivatives are neurotoxic, this disorder causes tremo r, psychosis, seizures, and cognitive dysfunction.
PHEOCHROMOCYTOMA
Clinical Presentation of Pheochromocytoma
- Symptoms
- Spontaneous severe anxiety: palpitations, sweating, panic
- Physical Exam Signs
- Tachycardia (Rapid heart rate)
- Hypertension (High blood pressure)
Biochemical Pathophysiology
As a simplification…
- Dopamine
- We can attribute the agitation and possible psychosis to the surge in Dopamine.
- Norephine & Epinephrine
- The sympathetic nervous system “fight or flight” symptoms relate to the surge in norepinephrine and epinephrine.
Laboratory Testing
- We test for pheochromocytoma in patients with unexplained episodic hypertension (high blood pressure) with blood and urine collection of:
- Catecholamine metabolite levels
- Metanephrine levels
- Specifically, we typically order:
- Urine and plasma free metanephrines
- Urine and plasma free catecholamines
- Urine homovanillic acid (HVA)
- Urine vanillylmandelic acid (VMA)
- The tests are highly sensitive, which leads to false positives. As anticipated, causes of false positives include:
- Sympathetic nervous system agitation (ie, psychophysiological stress)
- Exogenous triggers of catecholamines: pharmaceuticals, tobacco, caffeine, and illicit drugs.
Tumor Appearance
- Pheochromocytomas are black staining tumors (remember this was the color of melanin, another tyrosine derivative, as well) that classically grow out of the medullary layer of the adrenal gland.
- For a better understanding of the difference between the adrenal medulla and the adrenal cortex, see adrenal gland hormone production.
- Paragangliomas are essentially extra-adrenal pheochromocytomas
- They derive from cancerous autonomic nervous system tissue.
- True to the anatomy of the autonomic nervous system – head/neck ANS paragangliomas are parasympathetic whereas thorax and abdominal paragangliomas are sympathetic.
PHARMACOTHERAPEUTICS IN PARKINSON’S DISEASE
Parkinson’s disease (and for that matter, all Parksinonism syndromes) are Dopamine deficiency syndromes within the brain.
Carbidopa
- Indicate that the pharmaceutical carbidopa is used to block the decarboxylation of DOPA to dopamine, peripherally, and increase the bioavailability of dopamine centrally (in the central nervous system – where it is intended to treat Parkinson’s disease).
- Dopamine cannot cross the blood brain barrier but DOPA can, so Dopamine is administered systemically as L-DOPA. However, if it were administered without a decarboxylase inhibitor (such as carbidopa) it would be decarboxylated peripherally into Dopamine and patients would simply become nauseated.
- In the presence of a peripheral decarboxylase inhibitor, DOPA is taken up in the CNS and THEN decarboxylated to Dopamine (in the basal ganglia where it serves to replenish the deficient stores of Dopamine). Carbidopa (itself) doesn’t cross the blood brain barrier.
Therapeutics
- Levodopa: Dopamine Precursor
- So to treat a patient with Parkinson’s disease, let’s add levodopa as a Dopamine precursor.
- Carbidopa: DOPA decarboxylation Inhibitor
- At the same time, we need to add Carbidopa to block the peripheral DOPA decarboxylation of DOPA – we need to ensure that the levodopa makes it through the systemic circulation and enters the brain, otherwise it will simply act like any catecholamine within the periphery and increase blood pressure and heart rate but fail to impact the central nervous system Dopamine deficiency state.
- Ropinirole & Pramipexole: Dopamine agonists
- We can also add ropinirole or pramipexole, which are Dopamine agonists that optimize the release of Dopamine from the remaining Dopaminergic neurons within the substantia nigra.
- Entacapone: COMT Inhibitor
- We can add entacapone, which is a COMT inhibitor to increase the circulation of the Dopamine that we’ve stimulated or replaced (ie, we can inhibit catecholamine metabolism).
- Selegeline or Rasagaline: MAO-B Inhibitors
- And we can add selegeline or rasagaline, which are MAO-B (specifically) inihibitors which also increase Dopamine but via MAO inhibition (catecholamine metabolism inhibition).
- The B subunit is specific to Dopamine catalysis, whereas the MAO-A enzyme is less specific and also metabolizes norepinephrine and serotonin, thus drugs that inhibit MAO-A are potentially much more hazardous to use.
HYPERTHYROIDISM
Clinical Presentation
- Hypermetabolic state that manifests with:
- Weight loss, sweats, fevers, rapid heart rate.
- Skin and hair thinning
- Grave’s Ophthalmopathy
- Ocular protrusion and reddening
ALKAPTONURIA
- Show alkaptonuria, which we can think of as a melanin-like substance in the urine and joints.
Pathogenesis
- It occurs from a deficiency in homogentisate 1,2 dioxygenase.
- This results in a build-up of a melanin-like polymer called benzoquinone acetic acid (a product of the oxidation of homogentisic acid), which binds connective tissue and causes dark pigmentation or ochronosis (arthritis).
- Thus, we can think of alkaptonuria as the opposite of albinism + the build-up of acetic acid in the tissues irritates the joints and causes joint pain.
Presenting Symptoms
- Children: Dark Urine
- The presenting manifestation in children is typically urine that darkens when it sits for awhile (not common now with disposable diapers). The darkening occurs from the excess homogentisate in the urine (5,000 mg vs 20-30mg (normally).
- Adults: Join Pain
- In adults, the disease presents, typically, from joint pain from the build-up of acetic acid in the tissues, which irritates the joints.
TYROSINEMIA TYPE I
Pathogenesis: fumarylacetoacetate hydrolase deficiency
- Indicate that tyrosinemia type I (aka hereditary tyrosinemia, tyrosinosis) results from fumarylacetoacetate hydrolase deficiency.
Presenting symptom: “cabbage-like odor”
- Indicate that it characteristically causes a “cabbage-like odor” but importantly causes liver and kidney failure, polyneuropathy, and bone dysplasia (rickets), manifesting early-on with diarrhea, vomiting and tyrosine and its metabolites in the urine.
- Also consider that transient tyrosinemia (elevated blood levels of tyrosine) occurs in ~ 10% of newborns, most often due to vitamin C deficiency or immature liver enzymes due to premature birth.
Vesicular Budding and Fusion
STEPS OF VESICULAR BUDDING AND FUSION (+ PROTEINS INVOLVED)
- Cargo selection (cargo receptor, adaptor protein)
- Vesicular budding (adaptor proteins, coat proteins)
- Fission from donor membrane (dynamin)
- Vesicular coat dissociates
- Vesicular targeting and transport (Rab-GTPase, tethering protein)
- Fusion with target membrane (V-snare and T-snare)
PROTEINS OF VESICULAR BUDDING AND FUSION
- Cargo receptors – select and concentrate molecules to be transported in vesicle
- Adaptor proteins – bind cargo receptor and coat proteins
- Coat proteins – form protein scaffold around vesicle that facilitate facilitate vesicular budding
- Dynamin – GTPase involved in vesicular fission
- RabGTPase – associates with vesicle after coat has dissociated. Facilitates transport of vesicle to appropriate target membrane. Locks vesicle to target membrane by attaching tethering proteins
- Tethering proteins – anchored in target membrane, attach rabGTPase. Move vesicle close to target membrane for vesicular fusion.
- V-snares(vesicular) and T-snares(target membrane) – Play role in vesicular fusion
COAT PROTEINS DIRECT VESICLE TRANSPORT
- Clathrin +adaptin 1: Golgi → Lysosome
- Clathrin + adaptin 2: Plasma membrane → Endosomes (endocytosis)
- COP 1: Cis golgi → ER AND Later cisternae → Earlier ones (retrograde transport)
- COP II: ER → Cis golgi
