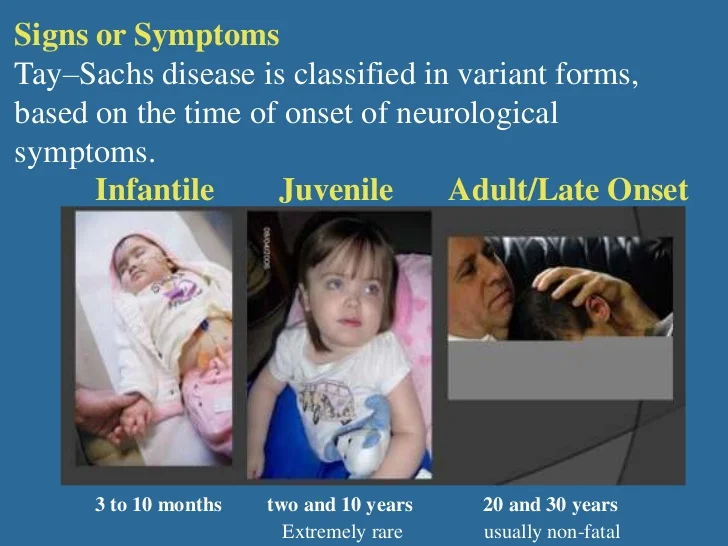
Autosomal recessive trait; due to lysosomal accumulation of sphingomylin resulting from inherited deficiency of sphingomyelinase.
The accumulations take place in spleen, liver, lungs, bone marrow, and brain.
A missense mutation causes complete deficiency of sphingomyelinase.
The enzyme deficiency block degradation of lipid, resulting in the accumulation of sphingomyelin within lysosomes in the macrophage-monocyte phagocyte system.
The following 6 types of Niemann-Pick disease have been described:-
Type A – Acute neuronopathic form
Type B – Visceral form
Type C – Chronic neuronopathic form
Type D – Nova Scotia variant
Type E – Adult form
Type F – Sea-blue histiocyte disease
Another classification divides this disease into –
Type A – acute infantile form.
Type B – less common, chronic, non – neurological form.
Type C – biochemically & genetically distinct form
Clinical Features:-
Niemann-Pick disease type A begins in the individual’s first few months of life. Symptoms include the following:
◦Feeding difficulties
◦Abdominal enlargement within 3-6 months
◦Progressive loss of early motor skills
◦Rapid decline leading to death by the time the patient is aged 2-3 years
Niemann-Pick disease type B is similar to Niemann-Pick disease type A, but the symptoms are more variable.
◦Abdominal enlargement may be detected in early childhood.
◦Respiratory infections recur.
◦No neurologic involvement is present.
Niemann-Pick disease type C usually affects school-aged children, but the disease may occur at any time from early infancy to adulthood.
Symptoms may include the following:
◦Unsteadiness of gait, clumsiness, problems in walking
◦Difficulty in posturing of the limbs
◦Slurred, irregular speech
◦Learning difficulties and progressive intellectual decline
◦Sudden loss of muscle tone, which may lead to falls
◦Seizures
Tremors accompanying movement.
Histologic features:-
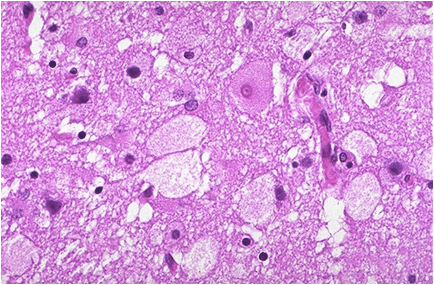
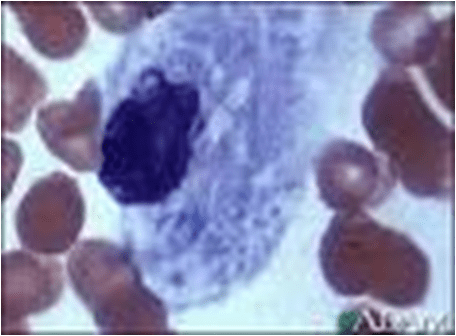
Niemann – Pick cells are foamy, lipid – laden cells distributed throughout RES.
Positive for cholesterol & only weakly positive for ALP.
Affected cell becomes extremely large, enlarged secondary to distention of lysosomes.
Treatment:-
Enzyme replacement therapy currently being explored.
Current treatment symptomatic; consists mainly of antibiotic therapy for infections of pulmonary involvement.
Clinical Features – Appears at 1 to 2 years of age; clear corneas, reduced intelligence, growth retardation, stiff joints
Differs from Hurler’s syndrome in –
Mode of inheritance (X – linked).
Absence of corneal clouding.
Milder clinical course..Results from deficiency of iduronate – 2 – sulfatase (I2S).
Without enough I2S, partially broken-down mucopolysaccharides accumulate in the organs and tissues of the body and become toxic.
Clinical features :-
Hunter syndrome is divided into two types.
I- Type A is he severe form, which usually is diagnosed in children aged 18-36 months.
Considered the classic form.
Children with type A may survive into the second and third decades of life.
Symptoms in type A may include:
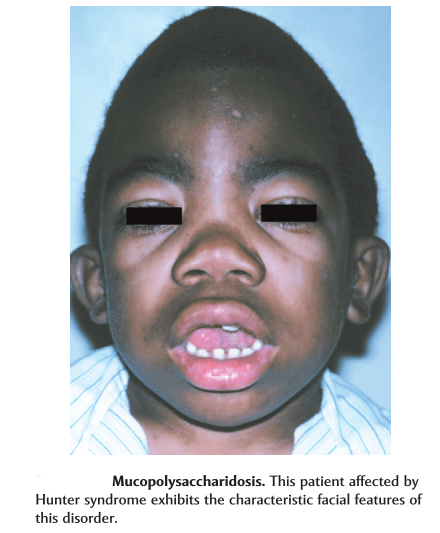
coarse facial features and short stature.
enlarged liver and spleen.
progressive and profound mental retardation.
ivory-colored skin lesions on the upper back and sides of the upper arms and thighs.skeletal changes, joint stiffness, short neck, broad chest, and too-large head.
progressive deafness.
atypical retinitis pigmentosa and visual impairment.
II.Type B Hunter syndrome is much milder than type A
May not be diagnosed until adulthood.
Individuals with type B may live into their 70s.
Their physical features are similar to those in type A.
Individuals with type B, however, usually have normal intelligence and do not have the severe skeletal problems of type A.
Diagnosis:-
In type A Hunter syndrome, the child’s appearance combined with other symptoms such as enlarged liver and spleen and the ivory-colored skin lesions can suggest the child has mucopolysaccharidosis.
Type B Hunter syndrome is much harder to identify, and might only be recognized when looking at the maternal relatives of a child with Hunter syndrome.
In either type, the diagnosis can be confirmed by a blood test for deficiency of I2S.
Treatment:-
Medical care is directed towards relieving the symptoms of Hunter syndrome.
Treatment with Elaprase (idursulfase) replaces I2S in the body and helps reduce symptoms and pain.
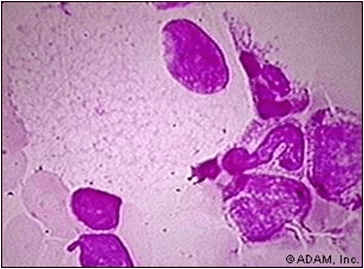
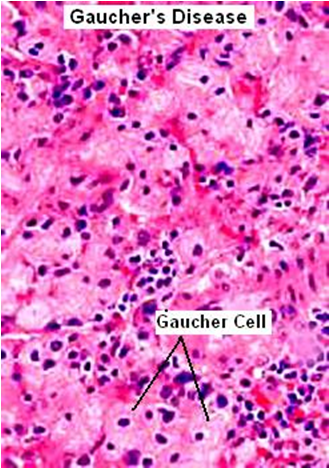
Excessive accumulation of intracellular mucopolysaccarides in many tissues & organs.
Accumulation of dermatan & heparan sulfate in cells of mononuclear – phogocyte system, fibroblasts & endothelial cells.
Affected cells are swollen, have clear cytoplasm resulting from accumulation of PAS +ve material in engorged, vacuolated lysosomes.
Involved fibroblasts assume appearance of ‘clear’ or ‘gargoyle’ cells.
‘Hurler cells’ relatively large with metachromatically staining cytoplasm with crescent shaped nuclei.
Cells not identified with normal H/E stain but with toluidine blue or Alcian blue/ aldehyde fuchsin stains.
Should be differentiated from mast cells.
Oral Manifestations:-
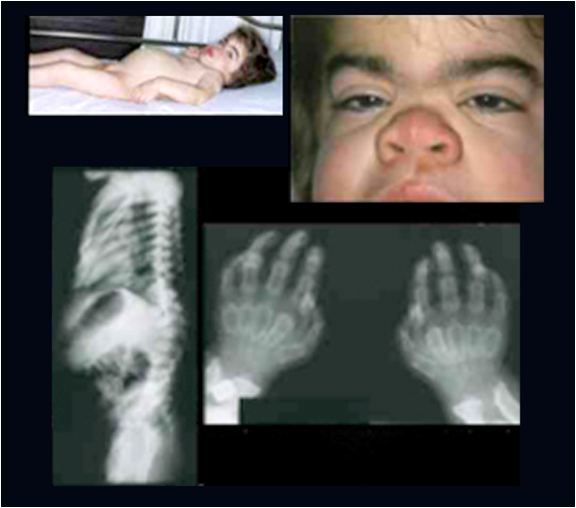
Shortening and broadening of the mandible with prominent gonions.
Localized areas of bone destruction in the jaws.
Teeth may be small and widely spaced.
Gingival hyperplasia has been repeatedly described in patients with Hurler syndrome.
the tongue is also characteristically enlarged.
Laboratory findings:-
Elevated levels of mucopolysaccharides in urine.
Metachromatic granules or ‘Reily bodies’ often demonstrated in cytoplasm of circulating lymphocytes.
Treatment:-
Because of multisystemic involvement in patients with mucopolysaccharidosis type I (MPS I), treatment is multidisciplinary and encompasses both the curative and palliative elements.
Corrective surgery may be necessary for patients with mucopolysaccharidosis type I (MPS I) who have joint contractures or foot and hand deformities.
Corneal transplants may be required if vision problems become severe.
Given the numerous mutations at this genetic locus, identification of which allele or alleles are involved requires referral to medical geneticists for diagnosis and genetic counseling.
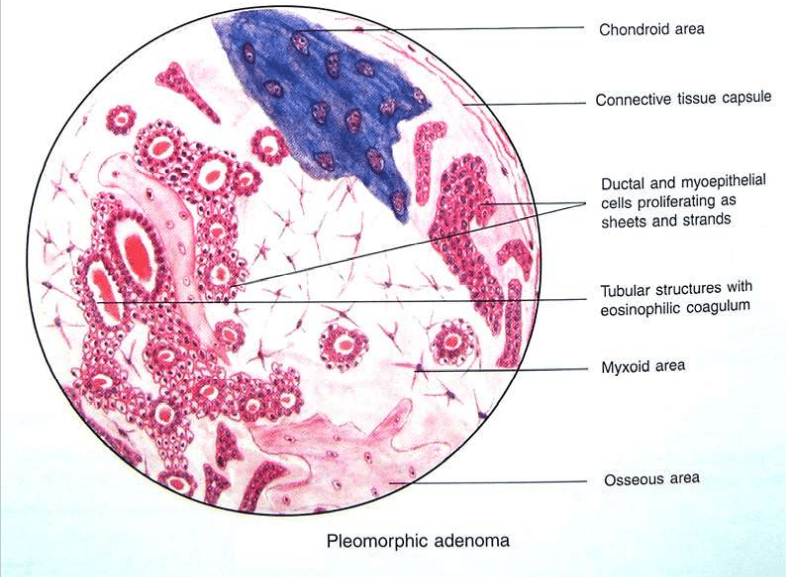
Name suggested by Willis. Most common neoplasm of salivary gland tumor. Benign neoplasm- consisting of cells exhibiting the ability to differentiate to epithelial (ductal and nonductal) cells and mesenchymal (chondroid, myxoid, osseous) cells. Other names:
Branchioma,
enclavoma,
teratoma,
cyindroma,
myxochondrocarcinoma.
Salivary gland tumor origin: EPITHELIAL
Shows cytogenic abnormalities in chomosomes- 12q13-15. Putative pleomorphic adenoma gene(PLAG1) has been mapped to chromosomes 8q12
Histogenesis:-
Currently, numerous theories centre around the myoepithelial cell and the reserve cell in the intercalated duct.
Ultrastructural studies have confirmed the presence of both ductal and myoepithelial cells in pleomorphic adenomas.
It follows that possibly either or both may play active roles in the histogenesis of the tumour.
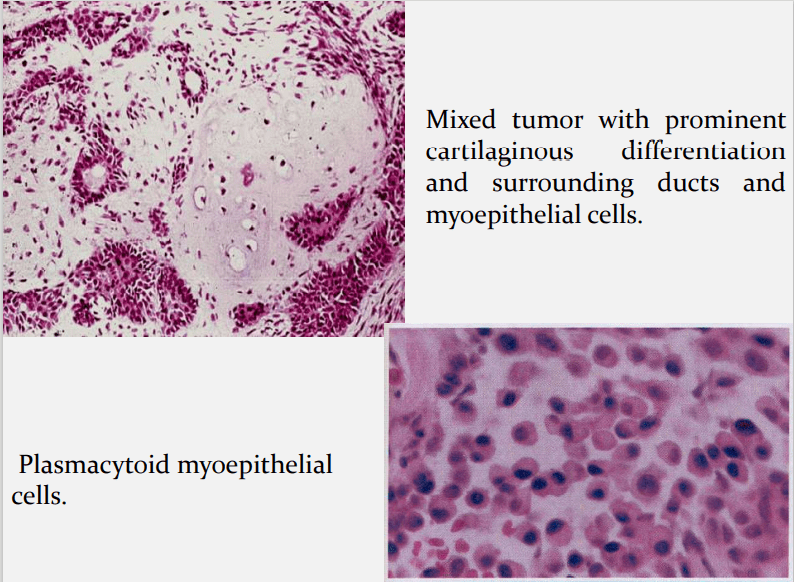
Hubner and his associates:- have postulated that the myoepithelial cell is responsible for the morphological diversity of the tumour, including the production of the fibrous, mucinous, chondroid and osseous areas.
Regezi and Batsakis:- postulated that the intercalated duct reserve cell can differentiate into ductal and myoepithelial cells, and the latter, in turn, can undergo mesenchymal metaplasia, since they inherently have smooth muscle-like properties. Further differentiation into other mesenchymal cells then can occur.
Batsakis:- has discussed salivary gland tumourigenesis, and while still implicating the intercalated duct reserve cell as the histogenetical precursor of the pleomorphic adenoma, stated that the role of the myoepithelial cell is still uncertain and that it may be either an active or a passive participant histogenetically.
Finally, Dardick and his associates have questioned the role of both ductal reserve and myoepithelial cells. They stated that a neoplastically altered epithelial cell with the potential for multidirectional differentiation might be histogenetically responsible for pleomorphic adenoma.
CLINICAL FEATURES:-
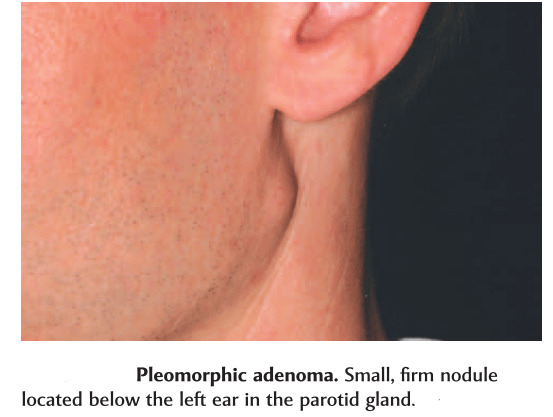
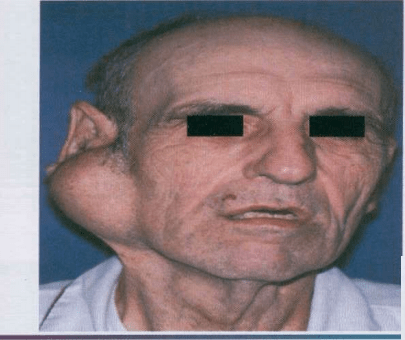
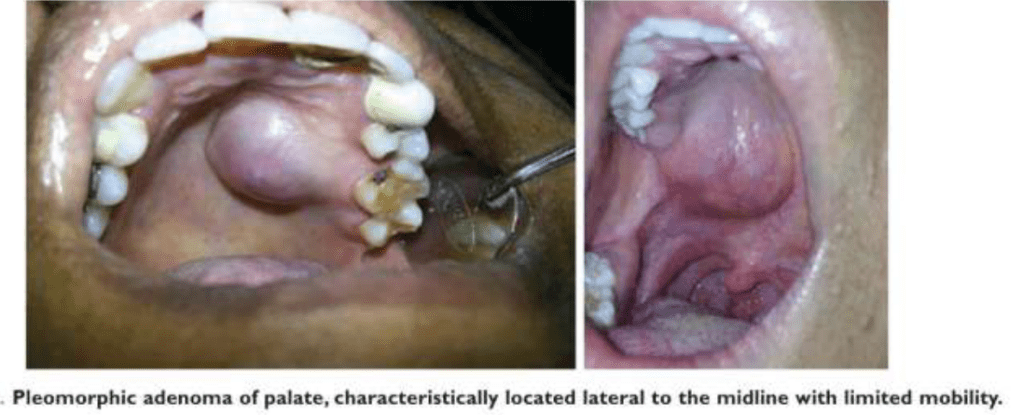
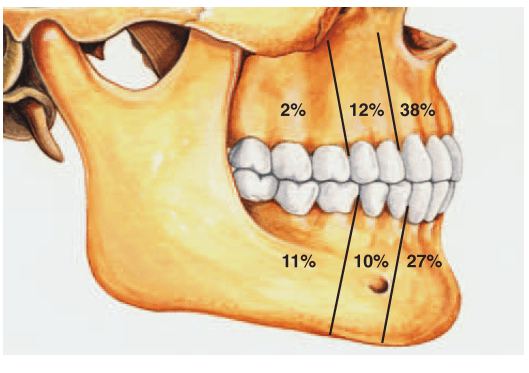
Most common tumor. Rate of occurance: 60-70%- parotid glands 40-60%- submandibular glands 40-70%- minor salivary glands seldomly- sublingual glands Age: 30-50 years Sex: female> male – 3:1 – 4:1 In parotid- presents in the lower lobe of the superior lobe as a mass over the angle of the mandible, below and infront of the ear.
Clinical presentation: painless, slow growing, firm mass, initially small in size and begins to increase in size. Initially movable but with continued growth become more nodular and less movable. Recurrent tumor- multinodular, fixed on palpation. Palate – intraorally common site. Seldom ulcerated- unless secondarily traumatized.
Slowly growing tumor of The parotid gland.
HISTOPATHOLOGY:-
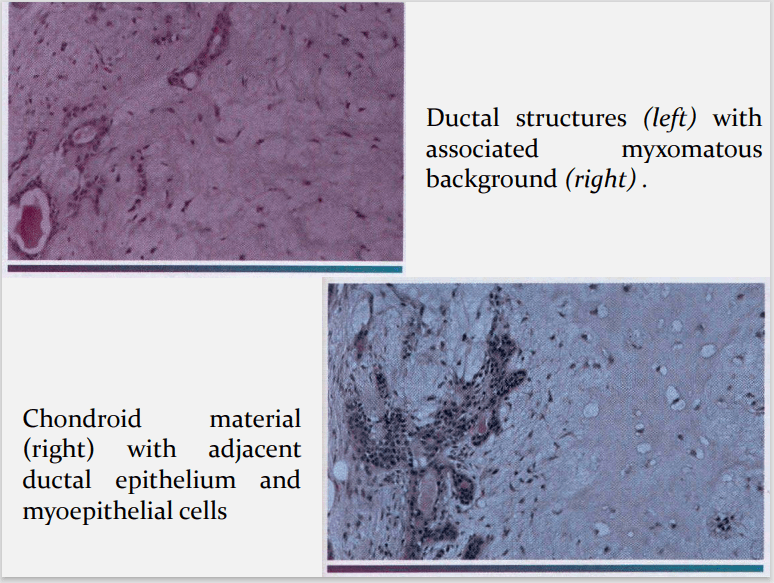
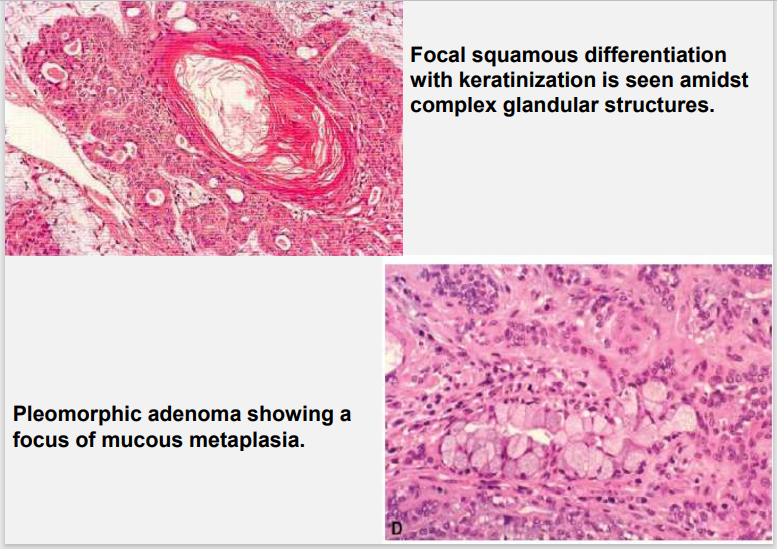
HALLMARK: Morphologic Diversity. Charecterized by- Variable, Diverse, Structural & histologic patterns. It demonstrate glandular epithelium and mesenchyme like tissue and the proportion of each component varies widely. Typically a well-circumscribed encapsulated tumor The epithelium often forms ducts and cystic structures or may occur as islands or sheets of cells , anastomosing cords and foci of Keratinizing squamous cells and spindle cells .
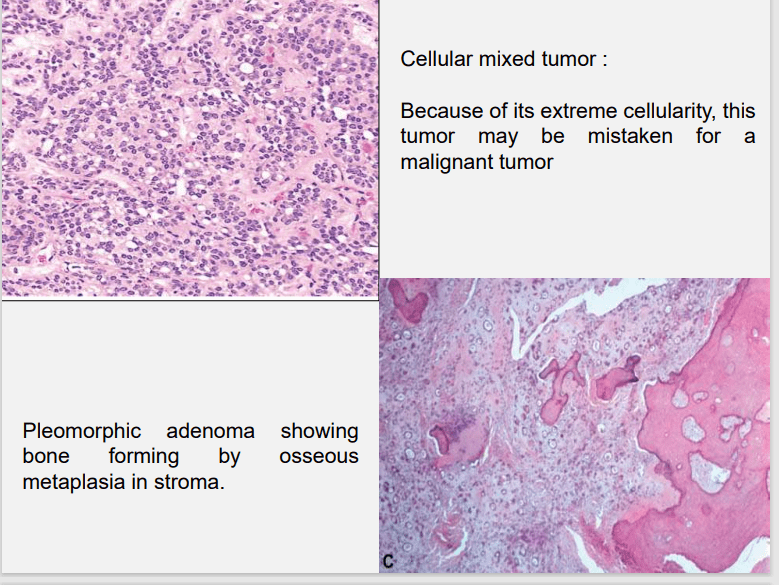
Foote and Frazell (1954) categorized PA into: a) Primarilly myxoid (36%) b) Myxoid and cellular component in equal proportions (30%) c) Predominantly cellular (22%) d) Extremely cellular (12%)
Myoepithelial cells are major component of PA. Have variable morphology- sometimes appearing as angular or spindled, some with eccentric nucleus resembling plasma cells. Are responsible for characteristic mesenchyme like changes. Vacuolar degeneration of myoepithelial cells can produce a chondroid appearance. the stroma exhibits areas of an eosinophilic hyalinized change, fat or osteoid also is seen.
Surgical excision Superficial parotidectomy with preservation of the facial nerve Local enucleation should be avoided – resulting in seeding of the tumor bed. Deep lobe of the parotid- total parotidectomy is usually necessary also with preservation of the facial nerve.
Submandibular tumors – Total removal of the gland with the tumor. Malignant degeneration is a potential complication, resulting in a carcinoma ex pleomorphic adenoma. The risk of malignant transformation is probably small, but it may occur in as many as 5% of all cases.
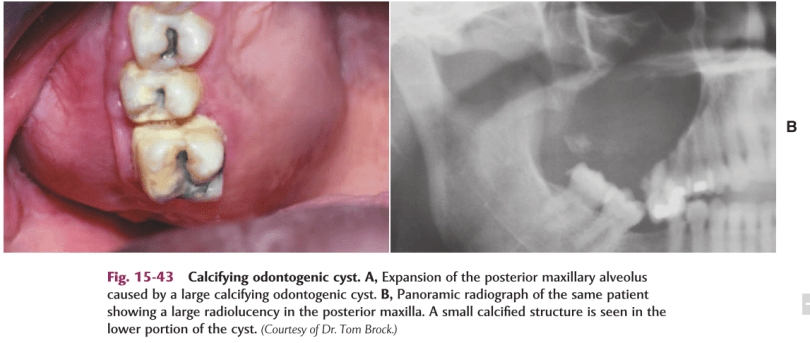
DEF:-Calcifying odontogenic cyst (COC), previously known as Gorlin cyst, is a rare, well-circumscribed, solid or cystic lesion derived from odontogenic epithelium that resembles follicular ameloblastoma but contains ‘ghost cells’ and spherical calcifications.
It Has many features of odontogenic tumor, therefore it is placed in the category of tumors in the latest WHO classification of odontogenic cysts and tumors.
In the latest WHO publication on odontogenic tumours (Prætorius and Ledesma-Montes, 2005) it was classified as a benign odontogenic tumour and was renamed calcifying cystic odontogenic tumour (CCOT).
CLINICALFEATURES:-
Age : Wide range, peak in 2nd decade.
Sex : Equal.
Site : Anterior segment of both jaws
Calcifying odontogenic cysts that are associated with odontomas tend to occur in younger patients, with a mean age of 17 years.
PATHOGENESIS:-
COC is a unicystic process and develops from the reduced dental epithelium or remnants of dental lamina.
The cyst lining has the potential to induce formation of dentinoid or even odontoma in adjacent CT wall.
CLASSIFICATION OF THE ODONTOGENIC GHOST CELLLESIONS:-
Group 1 : ‘Simple’ cysts Calcifying odontogenic cyst (COC)
Group 2 : Cysts associated with odontogenic hamartomas or benign neoplasms: calcifying cystic odontogenic tumours (CCOT).
Group 3 : Solid benign odontogenic neoplasms with similar cell morphology to that in the COC, and with dentinoid Formation
.Group 4 : Malignant odontogenic neoplasms with features similar to those of the dentinogenic ghost cell tumour Ghost cell odontogenic carcinoma.
SIGNS &SYMPTOMS:-
Swelling is the commonest complaint, seldom associated with pain.
Intraosseous lesions can cause hard bony expansion and resulting facial asymmetry.
Displacement of teeth can also occur.
RADIOLOGICALFEATURES:-
Intraosseous lesions produce well defined lucency which is usually unilocular.
Irregular calcified masses of varying sizes may be seen within the lucency.
Displacement of root/roots with or without root resorption and expansion of cortical plates also seen.
*Radiograph of a calcifying odontogenic cyst with well-demarcated margins extending from the right to the left premolar regions of the mandible. Numerous calcifications are present, some suggestive of small denticles.
HISTOLOGICALFEATURES:-
Lining is usually thin about 6 – 8 cell thick, may be thickened in other areas.
Lining shows characteristic odontogenic features with reversely polarized basal cell layer.
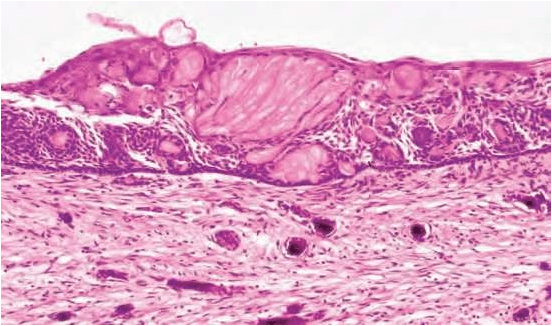
TYPICALLY – GHOST CELLS may be seen in thicker areas of lining.
Ghost cells are enlarged, ballooned, ovoid, eosinophilic cells with well
defined cell boundaries.
5.Some times many cells may fuse.
6.They represent abnormal keratinization and frequently calcify.
7.Tubular dentinoid and even complex odontome may be found in connective tissue wall close to epithelial lining.
Histological features of a calcifying odontogenic cyst with clusters of fusiform ghost cells and focal calcifications, lying in a stratified squamous epithelium.In this calcifying odontogenic cyst, there are sheets of ghost cells and a focal area in which there has been induction of a strip of dysplastic dentine (dentinoid).
DIFFERENTIALDIAGNOSIS:-
Based on radiographic appearance, following lesions must be included in the provisional diagnosis –
Rather this can be called a developmental anomaly.
It is characterised by heterotopic collection of sebaceous glands in various sites of the oral cavity.
It is said that the occurance of sebaceous glands in the mouth may be by inclusion in the oral cavity of the ectoderm.
This has some of the potentialities of skin during the development of the maxillary and the mandibular processes during the embryonic life.
Clinical features-
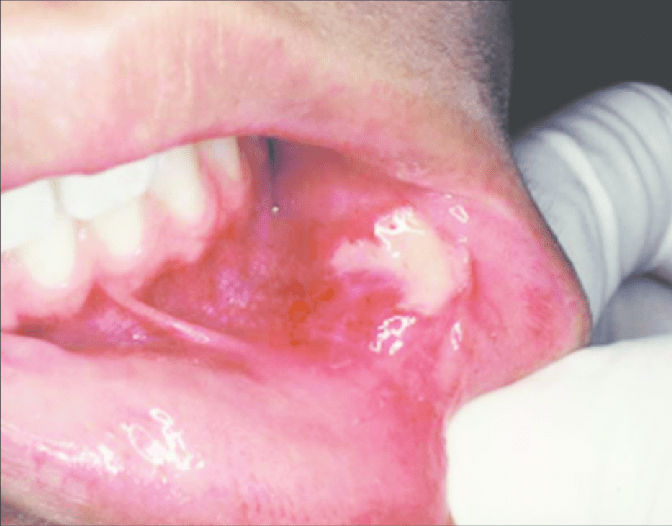
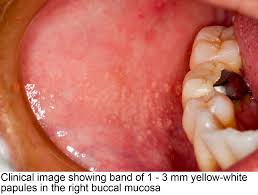
1.Appearance-
as yellow spots, seperated
or forming large plaques
project slightly above the suface of tissue
2. Site of appearance- found frequently in a bilateral symmetical pattern
mucosa of cheeks (opposite the molar teeth)
inner surface of lips
retromolar region
tongue
gingiva
palate
frenum
Besides the oral cavity they also appear in the oesophagus ,the female genital tract ,cervix uteri, male genitilia ,nipples, palms ,soles ,parotid ,larynx and the orbit .
3. Usually seen more in adults than children . This is due to the better development of sebaceous glands and hair system is not seen until puberty.
Histology
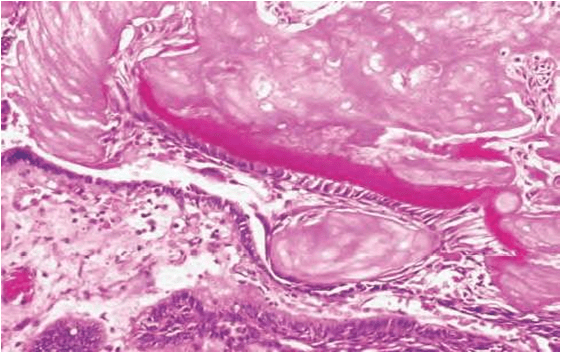
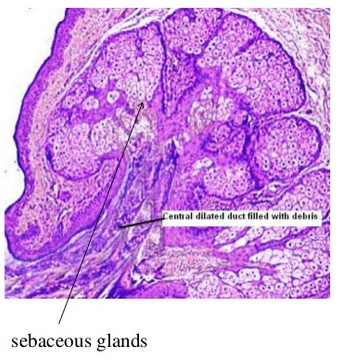
These are heterotopic collection of sebaceous glands and they are identical with those that are seen in the skin.
But they are unassociated with hair follicles and hair shaft from the gingiva. (this may be a very rare occurance )
Glands are located superficially.
There may be few or many lobules.
They are grouped around one duct or more ducts and they open at the surface of the mucosa .
The ducts may show keratin plugging.
Treatment-
It requires no treatment.
source – textbook of oral pathology shafers and google images .