Reiter’s syndrome is associated with urethritis, balanitis, conjunctivitis, and mucocutaneous lesions.
It is a disease of unknown aetiology, although there is evidence of an infec- tious origin.
It is one of the most common complications of non-specific urethritis and it clinically mimicks gonorrhoea, although the urethral discharge is negative for Neisseria.

CLINICAL FEATURES
>Reiter’s syndrome is more prevalent in young adult men, usually between 20 and 30 years of age.
>The male-to-female ratio is 9:1.
>There is a typical tetrad of manifestations: non- gonococcal urethritis, arthritis, conjunctivitis, and mucocutaneous lesions.
>Urethritis may be the first sign. The urethral discharge is usually associated with itching and burning sensation.
>The arthritis is often bilaterally symmetrical and usually polyarticular.
>Conjunctivitis is often so mild as to be overlooked.
>The skin lesions are similar to those seen in keratoderma blennorrhagica and consist of red or yellow keratotic macules or papules which eventually desquamate.
Oral Manifestations
Sites—it is seen on the buccal mucosa, lips and gingiva.
Oral lesions appear as painless, red, slightly elevated areas, some- times granular or even vesicular, with a white circinate border on the buccal mucosa, lips, and gingiva.
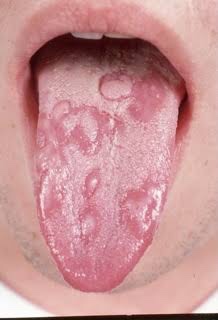
The palatal lesions appear as small, bright red purpuric spots, which darken and coalesce, while the lesions on the tongue closely resemble ‘geographic’ tongue.
Laboratory Findings
The patients usually have a mild leukocytosis, an elevated erythrocyte sedimentation rate, and pyuria.
- Differential Diagnosis
- • Geographic tongue and stomatitis—no skin changes, no visceral lesions are seen.
- • Pustularpsoriasis—Auspitz’ssignpresent.
• Behcet’ssyndrome—nourethritis,aphthaewithredhalo. - • Stevens-Johnson syndrome—acute appearance, moresevere clinical course, no arthritis or urethritis.
• Benign mucosal pemphigoid—blister formation, nourethritis, found in older patients.

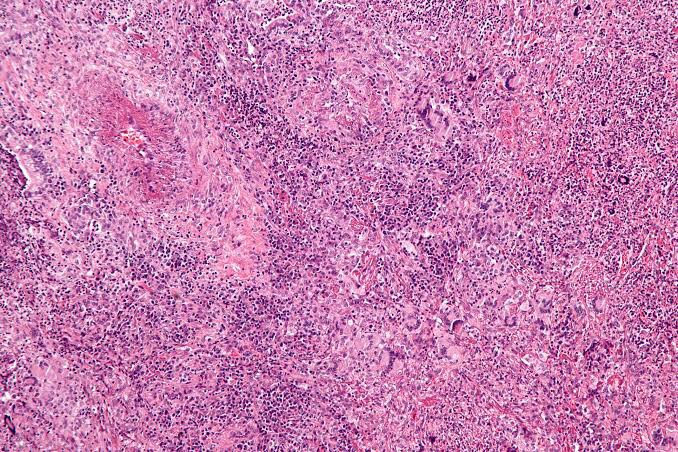
The microscopic findings are not diagnostic. They consist of parakeratosis, acanthosis, and polymorphonuclear leukocyte infiltration of epithelium, sometimes with mi- croabscess formation similar to psoriasis. The connective tissue shows a lymphocyte and plasma cell infiltrate.
- Management
- • Spontaneous remission—many patients undergo spontaneous remission.• Antibiotics—incasesymptomaticpatient,doxycycline or minocycline may be given.
- • Analgesics—nonsteroidalanti-inflammatorydrugsare given to manage arthritis.
- • Immunosuppressiveagents—immunosuppressiveagents like azathioprine and methotrexate are given in cases of most resistant cases.
REFERENCE- SHAFER’S TEXTBOOK OF ORAL PATHOLOGY [8TH ED} AND ANIL GHOM TEXTBOOK OF ORAL MEDICINE